N、S共掺杂碳点的制备及其对Cr6+的检测
时间:2022-12-08 14:20:03 来源:雅意学习网 本文已影响 人 
周叶红, 刘 竞, 刘 洋, 宋胜梅, 董 川
(山西大学环境科学研究所,太原 030006)
铬(Cr)是第四周期第VIB族金属元素,是人体必需的微量元素[1]。Cr的毒性受价态的影响,Cr3+对人体的身体健康有益;
Cr6+是重金属毒物,由于其强氧化性、致癌性和诱变性,可以将血红蛋白中的二价铁氧化为三价铁,使血红蛋白丧失携带氧的能力,同理,Cr6+也可使机体还原酶失去活性。铬中毒会引起皮肤红疹、水肿,鼻腔过敏以及胃炎、胃溃疡等症状[2]。铬污染主要来源于金属冶炼、皮革加工、染料生产等行业排放的含铬废水、废气及铬渣[3]。这些含铬污染物随着自然活动进入土壤、地下水、河流等,污染饮用水源和鱼类等海鲜制品。世界卫生组织(WHO)规定饮用水中的Cr6+的最大允许浓度为0.96 μmol/L[4-5],因此,为了保障人类健康,预防人类免受Cr6+的污染,环境中Cr6+的检测任务刻不容缓。目前Cr6+的国标法为高锰酸钾氧化-二苯碳酰二肼光度法,该方法有些预处理步骤繁琐、耗时长。因此,需要开发一种简单且经济高效的方法来有效测定Cr6+。
碳点(Carbon Dots,CDs)是一种新型的具有球形结构的准零维碳纳米材料,其表面具有丰富的官能团,这使得CDs具有出色的传感性能、易于功能化和良好的水溶性,利用碳点制备荧光探针具有高效、灵敏、快速检测实际水样中Cr6+的特点。目前已经有很多杂原子掺杂的碳点成功应用于水样中Cr6+的测定,如CDs/C3N4复合纳米探针[6]、Co-CDs[7]、D-甘露糖碳点[8]等。
本文开发了一种高灵敏测量和高线性范围的Cr6+的新型荧光探针,该探针可用于实际水样中Cr6+的检测,具有较好的应用前景。
1.1 试剂与仪器
试剂:分析纯亚硝基-2-萘酚-3,6-二磺酸钠,乙二胺,氢氧化钠,磷酸氢二钠,磷酸二氢钠,重铬酸钾,乙醇和金属氯盐购自上海阿拉丁试剂公司;
分析纯浓磷酸,抗生素和氨基酸购自美国Aldrich化学试剂公司;
超纯水为实验室自制(≥18.25 MΩ)。
仪器:JEM-2100高分辨率透射电子显微镜(日本电子株式会社JEOL),AXIS ULTRA DLDX射线光电子能谱仪(日本Kratos公司),TensorII傅里叶变换红外光谱仪(德国Bruker公司),vario ELCUBE元素分析仪(德国Elementar公司),FP-8300 FDP荧光分光光度计(日本分光株式会社JASCO),Lambda365紫外-可见吸收光谱仪(美国PerKinElmer公司),Eppendorf 5804离心机(德国Eppendorf公司),ZF-6紫外观察灯(上海嘉鹏科技有限公司),KQ-100E超声清洗器(江苏省昆山仪器有限公司),BGZ-76电热鼓风干燥箱(上海博迅实业有限公司),Scientz-18ND真空冷冻干燥机(上海博迅医疗生物公司)和1820c超纯水机(上海摩勒科学仪器有限公司)。
1.2 N,S-CDs的制备
取0.037 g 1-亚硝基-2-萘酚-3,6-二磺酸钠溶于20 mL超纯水中,加入500 μL乙二胺,将该混合物超声细散均匀后转入高温高压反应釜内衬中,在200℃的电热鼓风干燥箱中反应8 h。待水热反应结束后,所得溶液过滤、离心(8000 r/min,8 min),将上层清液转移至500~1000 Da的透析袋中透析8 h,再将透析后的淡黄色溶液冷冻干燥获得固体粉末,该固体粉末即所制得的N,S-CDs。
1.3 N,S-CDs的表征
通过透射电子显微镜(TEM)观察测定产物的形貌;
通过元素分析仪器对所制备N,S-CDs进行元素分析;
利用X-射线光电子能谱(XPS)和傅里叶变换红外光谱仪(FTIR)分析N,S-CDs化学结构及官能团组成;
最后通过紫外可见吸收光谱和荧光光谱对N,S-CDs的光学性质进行研究。
1.4 N,S-CDs的荧光量子产率测定
分别配制2 mL N,S-CDs溶液和2 mL硫酸奎宁溶液,将其转移至10 mm荧光比色皿中,然后上机测试。设置激发波长Ex为378 nm,通过控制加入荧光比色皿中样品溶液的浓度,控制紫外可见分光光度计的吸光度在0.01~0.1区间内,并使用紫外可见分光光度计测量溶液的吸光度。将荧光分光光度计的荧光积分区间设置为398~800 nm,用荧光分光光度计计算荧光积分面积。
荧光量子产率是衡量物质发射荧光能力的一个基本参数,其通常小于1。荧光量子产率越高,该物质的荧光越强,否则荧光越弱。相对荧光量子产率的计算公式如下[9]:

式中:Φ指量子产率;
n指荧光发射峰的积分面积;
A代表吸光度;
η指溶剂的折射率;
下标S和R分别指N,S-CDs溶液和硫酸奎宁溶液。硫酸奎宁的荧光量子产率为54%,在相同溶液中,ηS2/ηR2=1。
1.5 N,S-CDs对于Cr6+的荧光检测
取30 mg N,S-CDs粉末溶解于10 mL超纯水中,配置成浓度为3 mg/mL N,S-CDs储备液。
为了评估N,S-CDs的最佳浓度,在装有2 mL超纯水的10 mm比色皿中依次滴加10 μL N,S-CDs储备液,并测量其荧光强度。当加入320 μL N,S-CDs储备液时,荧光强度最大。再递加5 μL N,S-CDs储备液后,荧光强度逐渐降低。所以N,S-CDs溶液的最佳浓度为0.414 mg/mL。
为了评估N,S-CDs的pH稳定性,选择缓冲pH范围广的磷酸缓冲溶液(Phosphate Buffer,PBS)为本试验的缓冲溶液。测量并记录N,S-CDs在pH值为2~13磷酸缓冲溶液中荧光强度的变化,考察pH值对N,S-CDs溶液稳定性的影响。为了获得N,S-CDs的选择性,需要将21种终浓度为0.43 mmol/L的金属离子(Na+、Mg2+、Al3+、K+、Ca2+、Ti3+、Mn2+、Ba2+、Fe2+、Cr6+、Fe3+、Cd2+、Co2+、Ni2+、Cu2+、Ag+、Cd2+、Hg2+、Sn2+、Zn2+、Pb2+),14种终浓度为0.43 mmol/L的阴离、Cl-、Br-、I-),17种终浓度为0.043 mmol/L氨基酸(天冬氨酸(Asp)、谷氨酸(Glu)、苏氨酸(Thr)、缬氨酸(Val)、酪氨酸(Tyr)、脯氨酸(Pro)、苯丙氨酸(Phe)、半胱氨酸(Cys)、丙氨酸(Ala)、异亮氨酸(Ile)、甘氨酸(Gly)、色氨酸(Trp)、蛋氨酸(Met)、组氨酸(His)、亮氨酸(Leu)、丝氨酸(Ser)、赖氨酸(Lys))和14种终浓度为0.043 mmol/L抗生素(阿奇霉素(AZM)、克拉霉素(CLA)、卡那霉素(Kana)、万古霉素(VAN)、甲砜霉素(THI)、庆大霉素(GEN)、四环素(TCY)、头孢拉定(CH)、青霉素G钠(PG)、熊果酸(UA)、奥硝唑(ORN)、齐墩果酸(OA)、替硝唑(TIN)、甲硝唑(MET)),加入到2 mL N,S-CDs溶液中,充分摇匀,记录其荧光强度。
为了获得N,S-CDs的抗干扰性能,将响应离子10倍浓度的干扰离子加入2 mL N,S-CDs溶液,充分摇匀,记录其荧光强度。再将响应离子加入干扰离子与N,S-CDs的混合溶液中,充分摇匀并记录其荧光强度。
对于Cr6+的检测,配制一系列不同浓度(0~336 μmol/L)的Cr6+溶液,并加入到2 mL N,S-CDs溶液(4.14 mg/mL)中,充分摇匀60 s,设置荧光分光光度计的工作条件为λEx=378 nm,λEm=467 nm,激发和发射狭缝分别为10 nm,上机测量并记录其荧光强度,得到一系列的荧光猝灭谱图,然后进行线性范围和检测限的计算。
1.6 N,S-CDs用于环境水样中Cr6+检测
取实验室自来水和太原市令德湖水为实际水样。将实际水样离心过滤煮沸备用。通过荧光光谱法测试实际水样中Cr6+的含量,然后做Cr6+的加标回收试验,测试Cr6+的加标回收率。
2.1 N,S-CDs的结构表征
从TEM透射电镜图(图1(a))可以看到,N,SCDs为均匀分散的球形碳纳米颗粒,尺寸均匀分布在8~12 nm之间,通过100个粒子的统计测量,计算其平均粒径为10.31 nm(图1(b))。然后分析N,S-CDs的元素含量。N,S-CDs由C、H、N、O和S组成,各元素分别占总质量的37.67%、6.5%、21.36%、27.49%和6.98%(计算值),经计算所制备N,S-CDs的经验式为C31H65O17N15S2。

图1 N,S-CDs的TEM图像和尺寸分布
利用傅里叶变换红外光谱仪(FTIR)和X射线光电子能谱仪分析N,S-CDs化学结构及官能团组成。FTIR光谱见图2(a),N,S-CDs在3480~3314 cm-1及1500 cm-1处的吸收峰是N—H的伸展振动的特征;
3015~3019 cm-1处的宽吸收峰属于不饱合C—H伸展振动;
3000~2800 cm-1处的宽吸收峰属于饱合C—H伸展振动;
2548 cm-1为杂环中C—S的典型伸展振动;
2193 cm-1处的吸收峰属于N N的伸展振动;
3000 cm-1为中心宽峰,属于—COOH的—OH伸展振动;
1636 cm-1处的吸收峰属于—COOH的C O伸展振动;
1580和1490 cm-1处的吸收峰属于碳点六元环C—C骨架振动;
1350 cm-1处的宽吸收峰属于NO2的伸缩振动;
1160和1037 cm-1处的宽吸收峰分别属于羟基C—O不对称伸展振动和对称伸展振动;
820和680 cm-1处的尖锐吸收峰为碳点骨架上C—H弯曲振动。这些FTIR吸收峰说明N,S-CDs已被N—H、N N、C—S、C O等官能团功能化了,从而赋予N,S-CDs良好的水溶性、光学性能和优异的传感性能。
XPS全谱在161.45、281.72、396.33、529.89 eV处有4处特征峰,分别对应于S2p、C1s、N1s、O1s(图2(b))。图2(c)为高分辨的C1s谱图,高分辨的C1s可分解为3个峰,分别对应于285.1 eV(C—C)、286.4 eV(C O、C—O)、288.6 eV(C—N、C—S)。N1s的XPS谱图有两个含氮官能团,包括吡啶氮(399.4 eV)和氧化氮(401.3 eV)(图2(d))。O1s谱可分解为2个特征峰,分别对应于531.5 eV(O—H、S O、N—O)和533.2 eV(C—O、C O)(图2(e))。S2p可分解为2个峰,分别是168.0 eV、169.0 eV(图2(f))。N,S-CDs表面含有丰富的、N—H、—COOH,大大增强了N,S-CDs的水溶性、光稳定性和选择性。基于上述元素分析、FTIR和XPS数据,表明N、S原子已被掺杂到N,S-CDs中。

图2 FTIR和XPS对N,S-CDs进行官能团表征
2.2 N,S-CDs的光谱性能
如图3(a)的紫外吸收光谱所示,N,S-CDs在240和250 nm处具有明显的吸收峰,这可能是C C键的π-π*跃迁引起的,276 nm处的吸收峰可能是由于C O键n-π*跃迁引起的,380 nm处的吸收带可能是由于CDs外部的官能团吸收引起的。
图3(a)插图说明碳点溶液在可见光下为透明淡黄色溶液,紫外灯下发出明亮的蓝色荧光。荧光光谱中,在激发和发射狭缝分别为10 nm的工作条件下,测得N,S-CDs的最佳激发波长λEx为378 nm,而最佳发射波长λEm=468 nm(图3(a))。由于N,S-CDs的最佳激发波长(λEx=378 nm)与硫酸奎宁的最佳激发波长(λEx=360 nm)相近,故选择利用硫酸奎宁溶液为参比测定N,S-CDs的荧光量子产率。通过量子产率公式计算出N,S-CDs的相对荧光量子产率约为3.0%。通过调节N,S-CDs激发波长(328~428 nm),N,S-CDs的发射峰位置改变,说明N,S-CDs具有激发波长依赖性(图3(b))。
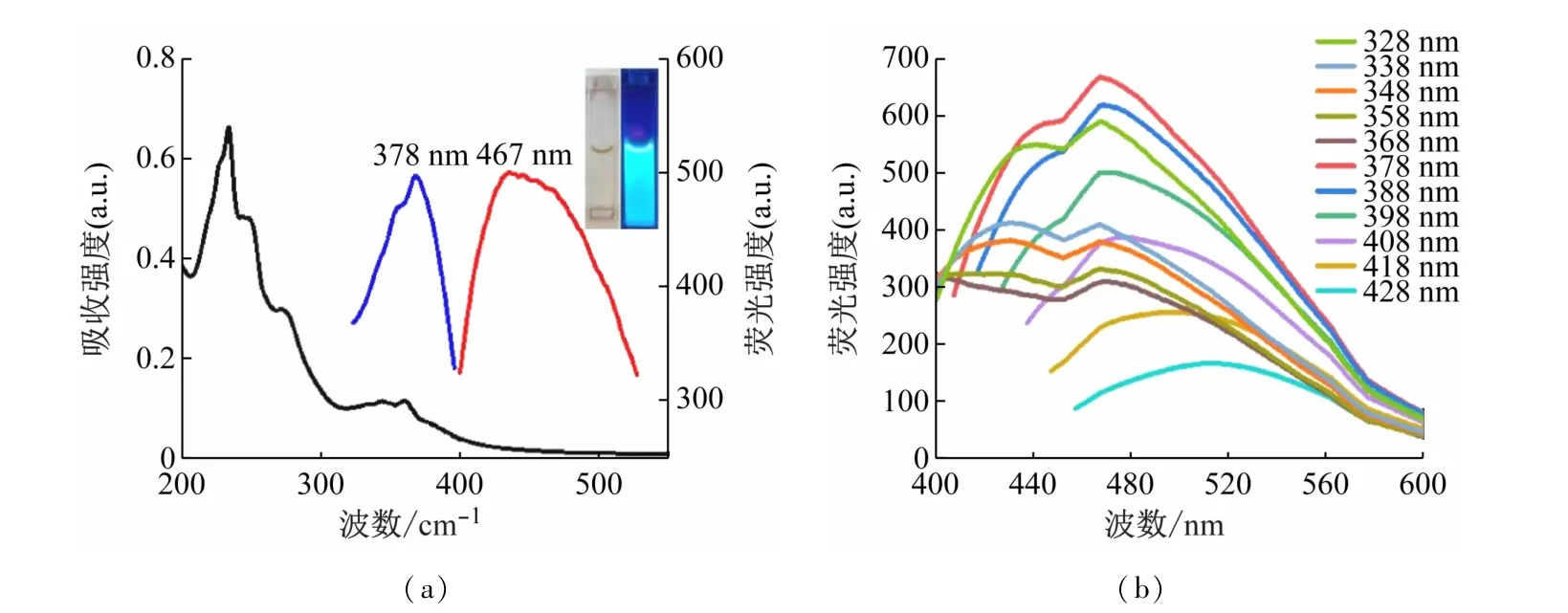
图3 (a)N,S-CDs的紫外可见吸收光谱(黑线),荧光激发(蓝线)和发射(红线)。插图:N,S-CDs在自然光(左)和紫外线(右)下的拍摄图。(b)N,S-CDs在不同激发波长(328~428 nm)下的发射光谱图
2.3 N,S-CDs对于Cr6+的荧光检测
为了评估N,S-CDs探针的选择性,在N,S-CDs溶液中分别加入了21种金属离子、14种阴离子、17种氨基酸和14种抗生素,结果如图4所示。
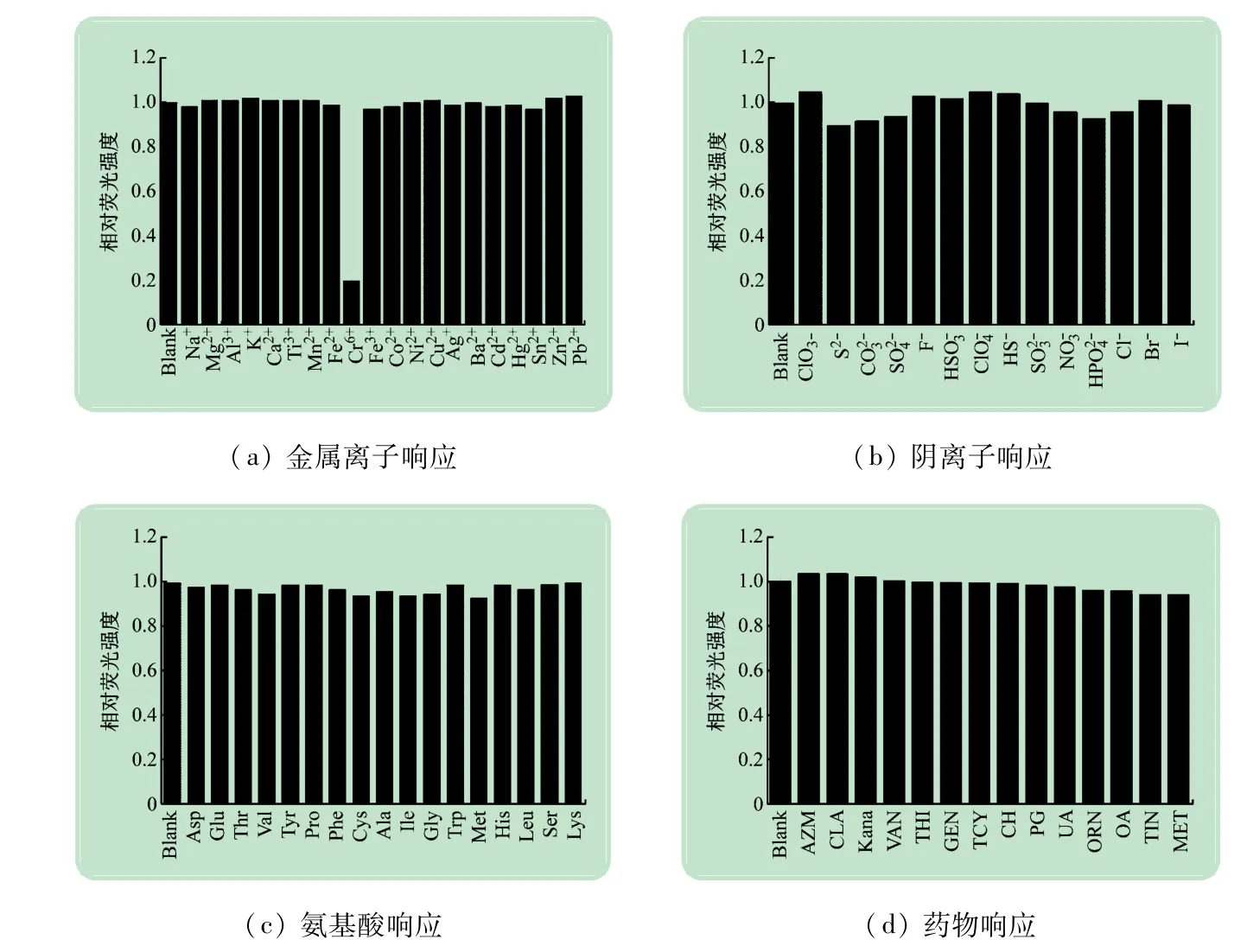
图4 N,S-CDs响应图谱
N,S-CDs的荧光强度在Cr6+溶液中明显降低了,Cr6+可以显著猝灭N,S-CDs的荧光强度,且抗生素、氨基酸、阴离子和其他金属离子对N,S-CDs荧光强度的影响甚微,可忽略不计。所以N,S-CDs对Cr6+有专一的选择性,可以将N,S-CDs作为Cr6+的无标记“开-关”荧光传感探针。
N,S-CDs检测Cr6+的抗干扰实验如图5所示,原N,S-CDs溶液(蓝)的相对荧光强度为1,加入4.3 mmol/L金属离子和阴离子后(红),N,S-CDs溶液的荧光强度减弱了10%左右,再添加0.43 mmol/L Cr6+后(绿),荧光强度明显猝灭了80%左右,与单独加入Cr6+时的荧光猝灭强度基本一致,证明了Cr6+与其他离子共存时不会对荧光产生明显的干扰,为N,S-CDs在复杂体系实现对Cr6+的检测奠定了理论基础。

图5 不同金属离子和阴离子对N,S-CDs的荧光强度的影响
由图6可知,N,S-CDs在超纯水体系中稳定,且荧光猝灭效果好,故选择超纯水体系为N,S-CDs的检测体系。
图7(a)为滴定不同浓度的Cr6+时N,S-CDs荧光猝灭图。由图7(a)可知,N,S-CDs的荧光强度随着Cr6+的不断加入而降低。从7(b)可以看出,Cr6+对N,S-CDs的荧光猝灭可以用两段线性范围来拟合。在2~100 μmol/L范围内,Cr6+对N,S-CDs荧光强度猝灭呈线性,线性拟合方程为:

检出限为68.23 nmol/L(图6(c));
在100~330 μmol/L范围内,线性拟合方程为:F0/F=0.04733c(Cr6+)-2.08657,R2=0.991(图7(d)),检出限为206 nmol/L,F0和F分别为猝灭前后N,S-CDs的荧光强度。

图6 添加Cr6+对N,S-CDs荧光强度的影响

图7 (a)不同浓度Cr6+对N,S-CDs荧光光谱的影响;
(b)、(c)、(d)F0/F与Cr6+浓度(0~350 μmol/L)的关系图
2.4 Cr6+检测方法的性能比较
将本试验所构建的Cr6+荧光探针N,S-CDs与其他CDs探针进行检测极限及荧光性能等方面的比较,结果如表1所示。本试验合成了蓝色荧光碳量子点(N,S-CDs),确定了两条Cr6+检测的线性范围,分别是2~100 μmol/L和100~330 μmol/L,最低检出限分别为68.23 nmol/L和206 nmol/L,远低于世界卫生组织(WHO)规定饮用水中的Cr6+的最大允许浓度(960 nmol/L),可以应用于环境水样中Cr6+的检测。

表1 基于CDs荧光探针检测Cr6+的方法比较
2.5 N,S-CDs测定Cr6+的检测机理
图8显示了N,S-CDs与不同浓度Cr6+(0~216.5 μmol/L)的紫外吸收光谱图,N,S-CDs的n-π*跃迁和π-π*跃迁没有吸收峰位置的变化,说明N,S-CDs与Cr6+之间没有形成新的化合物。在340~380 nm之间紫外吸收峰显著升高,而N,S-CDs的激发波长也位于这一范围,所以可能是Cr6+吸收了N,S-CDs的激发光能量,基于Cr6+的内滤效应[13-15],导致了N,S-CDs的荧光猝灭。
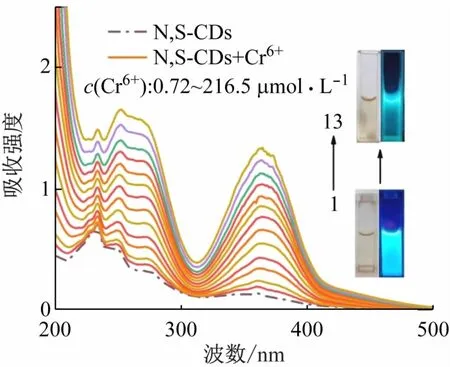
图8 不同浓度Cr6+(0~216.5 μmol/L)存在下N,S-CDs的紫外吸收光谱(插图;
加入216.5 μmol/L Cr6+的N,S-CDs溶液(上)和N,S-CDs(下)在日光和紫外灯下的拍摄图)
2.6 实际样品中Cr6+的检测
实际水样中Cr6+检测分析如表2所示,实际水样中Cr6+的加标回收率为95.3%~103.8%,RSD≤4.38%。这些数据表明,试验所合成的新型荧光碳量子点N,S-CDs可以作为一种高效灵敏的“开-关”型荧光探针,应用于Cr6+的检测。
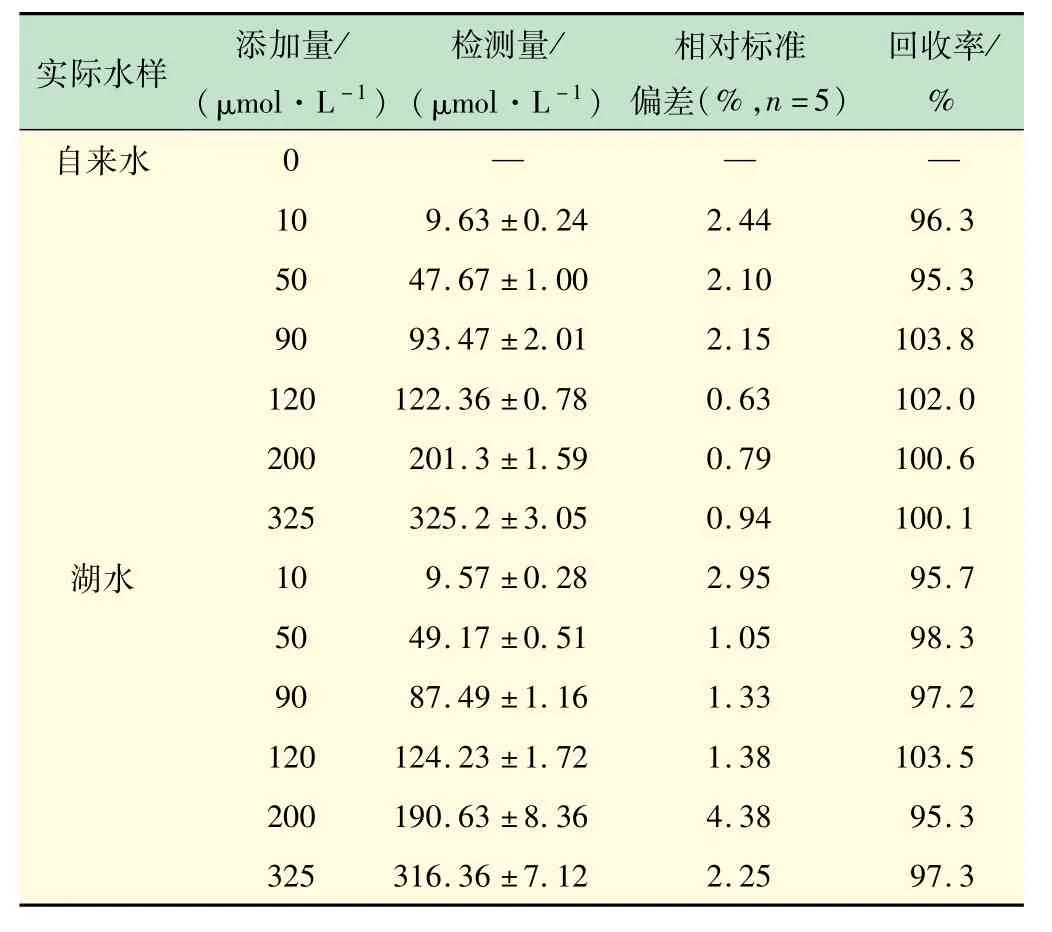
表2 实际水样中Cr6+的测定
通过一步水热法,以乙二胺和1-亚硝基-2-萘酚-3,6-二磺酸钠为原料,制备了具有蓝色荧光的新型氮硫掺杂碳点(N,S-CDs)。该N,S-CDs具有优良的荧光性能和选择性,并可作为Cr6+的免标记的荧光传感探针,线性范围为2~100 μmol/L和100~330 μmol/L,最低检出限为68.23 nmol/L。该N,S-CDs可以快速、准确的检测水样中的Cr6+,可以应用于环境监测。
猜你喜欢 探针水样荧光 荧光探针在游离肼检测中的研究进展当代化工(2020年2期)2020-03-18磁性四氧化三铁氮掺杂石墨烯磁性固相萃取测定水样中的6种醛酮化合物分析化学(2019年3期)2019-03-30Fenton与超声空化联合技术深度处理炼油废水的研究环境与发展(2018年6期)2018-09-17魔力荧光色小资CHIC!ELEGANCE(2018年28期)2018-09-14玛卡荧光碳点的合成及用于苦味酸的荧光检测分析化学(2016年12期)2017-02-04Fluorescence world荧光人间小资CHIC!ELEGANCE(2016年15期)2016-07-26水货博客天下(2014年9期)2014-09-03通过接触测试来提高探针痕迹的一致性卷宗(2014年7期)2014-08-27并行测试探针卡的移动规则选择卷宗(2014年1期)2014-03-20DNA探针在高中生物学教学中的应用中学生物学(2008年12期)2008-12-27